To maintain homeostasis, biological systems require a careful balance between diverse cellular components and their interactions. Disease states, but also therapeutic interventions, can be understood as perturbations of this system, either driving it away from homeostasis, or aiming to restore it, respectively. The overarching ambition of this project is to identify key principles that govern how combinations of internal and external perturbations converge into a combined response, thanks to network biology.
“Every day, millions of people are taking medications that will not help them. The top ten highest-grossing drugs in the United States help between 1 in 25 and 1 in 4 of the people who take them […]. There are even drugs that are harmful to certain ethnic groups because of the bias towards white Western participants in classical clinical trials.”
– N.J. Schork (Nature, 2015, doi:10.1038/520609a)
We propose a combined theoretical and experimental approach for the construction of a high-resolution directed interaction map of context-dependent drug response. Specifically, we will quantify the changes that chemical compounds and gene knockouts induce in cell morphology. Comparing the effects of genes and drugs individually with the effect of their combination allow us to identify interactions between them. By systematically mapping out and characterizing these interactions in a large-scale combination screen we expect to (i) identify rules for the emergence of drug-gene interactions, (ii) obtain new insights into cell-type specific responses and (iii) deepen our understanding of how different perturbations are processed by the molecular network of the cell.
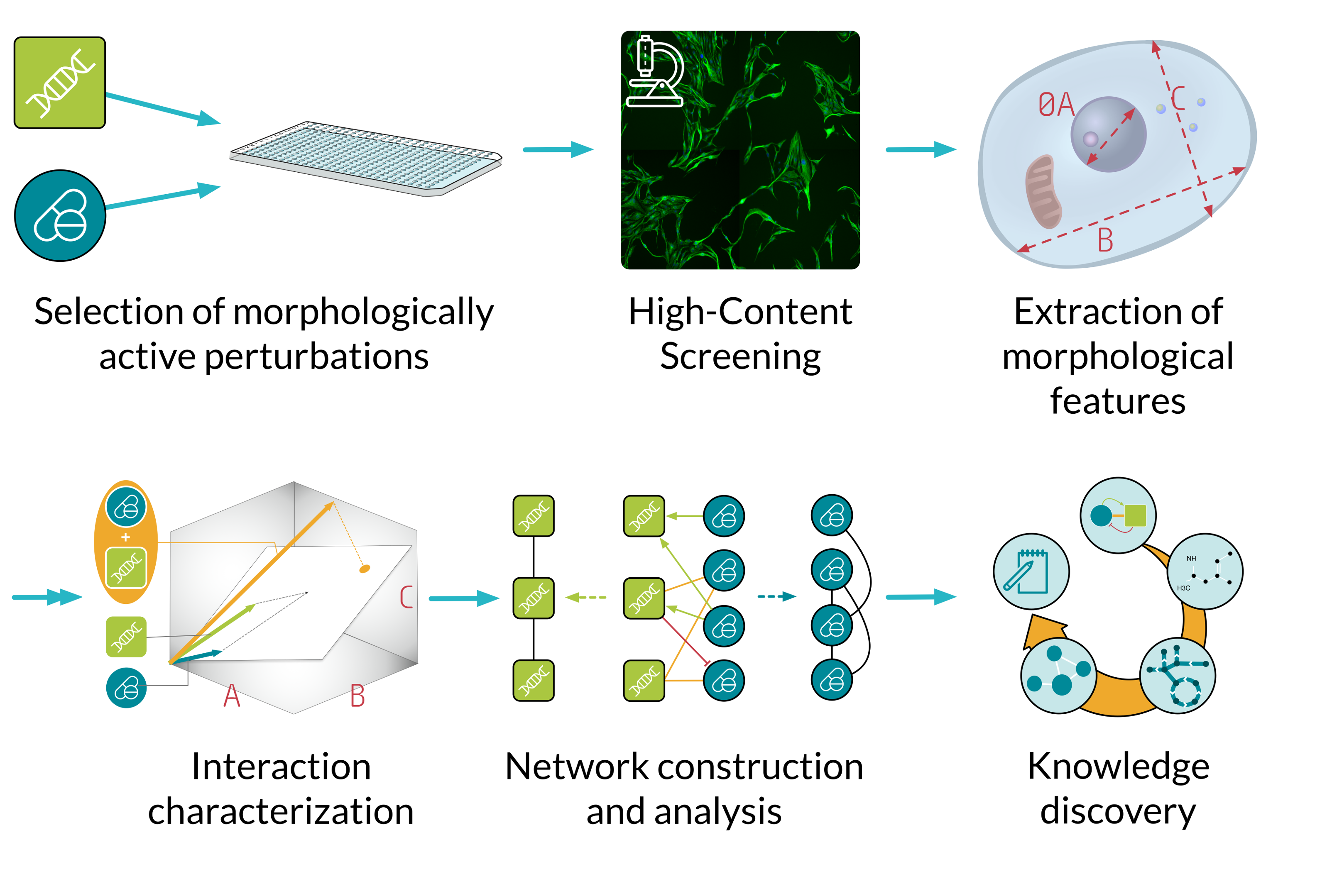
This ongoing screen is performed in MCF10A, a human epithelial cell line. For all pairwise combinations between 311 compounds and 200 CRISPR knockouts, microscopy images will be processed and analyzed in order to extract morphological features describing the cell. Using a novel mathematical framework developed previously in the Menche lab, we obtain a detailed landscape of directed interactions between internal and external perturbations. Finally, we will characterize and interpret the resulting map using diverse molecular and phenotypic datasets, ranging from functional gene annotations to known disease associations.
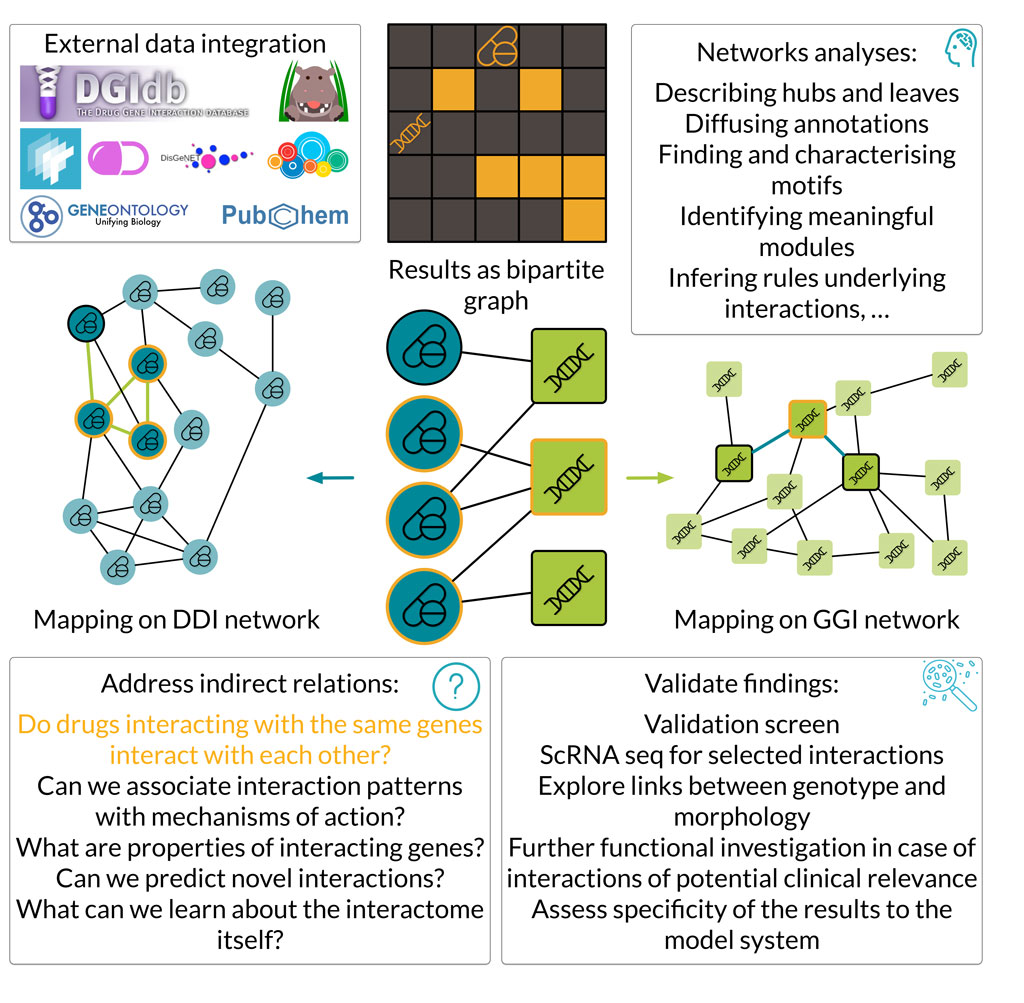
This approach harnesses the power of systems biology and network science to go beyond what can be concluded from the interactions individually, thus allowing for system-wide conclusions.